Characterization of a Mouse Model of Rett Syndrome
March 31, 2025
Rett Syndrome is a genetic neurological disorder caused by X-linked Mecp2 gene mutations. Males are more strongly affected and usually die before birth or early in life. Rett Syndrome symptom onset is 6-18 months of age with impairments including: seizures, severe mobility loss, language impairment, sleep challenges, Autistic-like symptoms and cognitive deficits.
PsychoGenics employs the Mecp2 female heterozygous and Mecp2 male knockout mouse model (Mecp2tm1.1Bird) as a preclinical model of Rett Syndrome.
Mecp2 Mouse Model Differences in Molecular Biomarkers
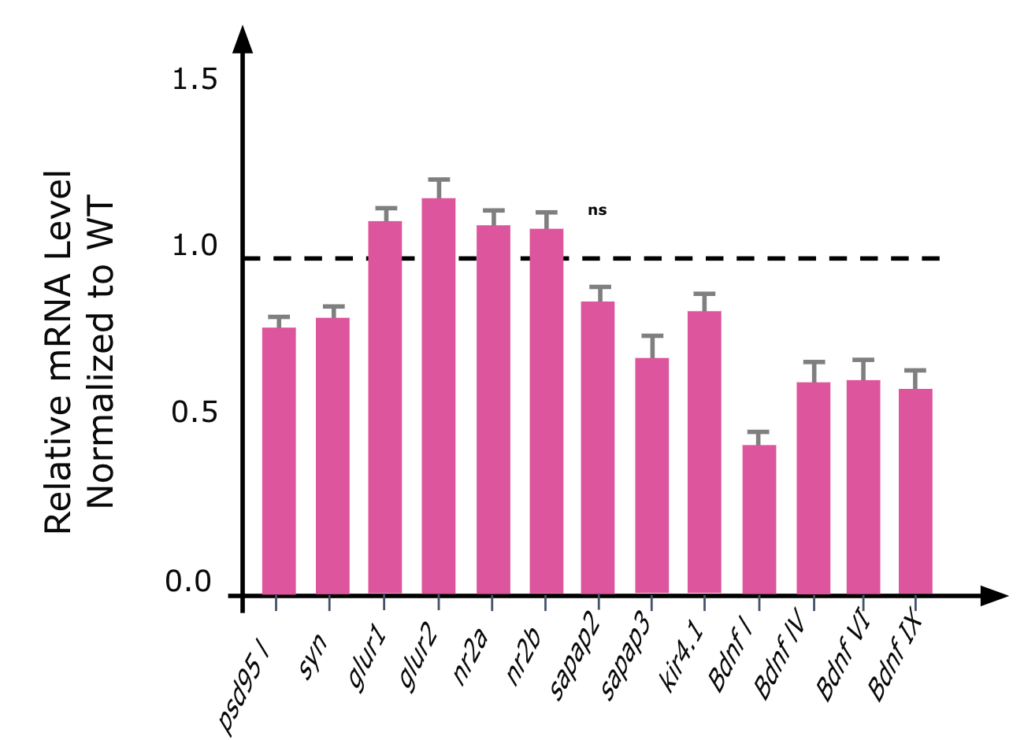
Female Mecp2 HET mice (4 months old) showed decreased levels of PSD95, Synaptophysin, Sapap3, Kir4.1, and increased levels of glutamate receptor subunits, as well as decreased levels of BDNF isoforms.
Deficits in Visual Acuity
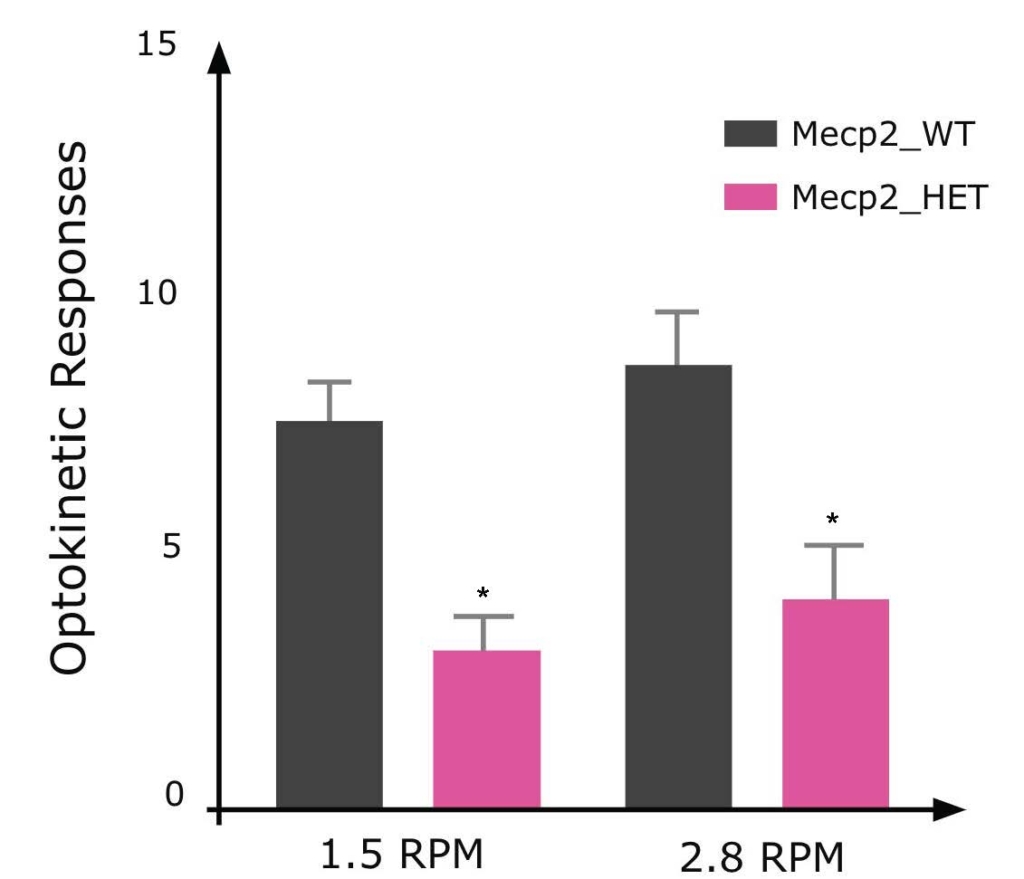
Visual performance when tracking a moving object is impaired in female Mecp2 HET mice resulting in fewer correct optokinetic responses compared to WT mice.
Deficits in Fear Conditioning
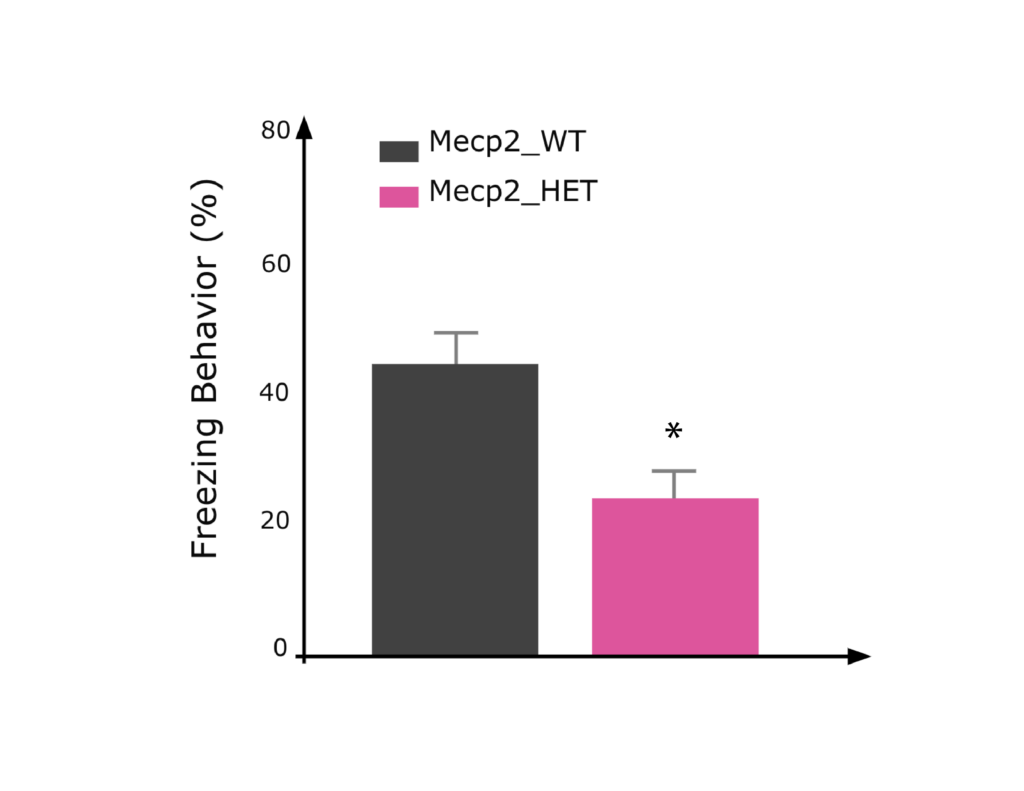
Female Mecp2 HET mice showed decreased freezing during contextual fear conditioning test compared to WT mice.
Changes in Respiration
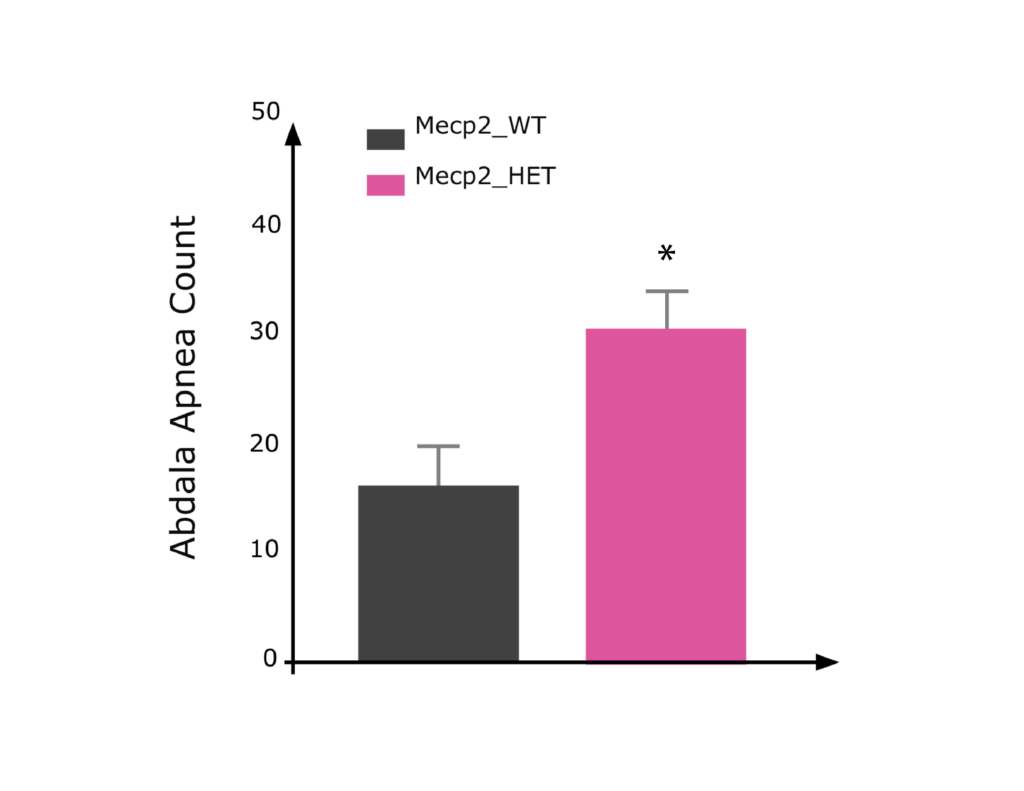
Female Mecp2 HET mice showed elevated apnea counts relative to WT mice when tested in a whole-body plethysmograph system.
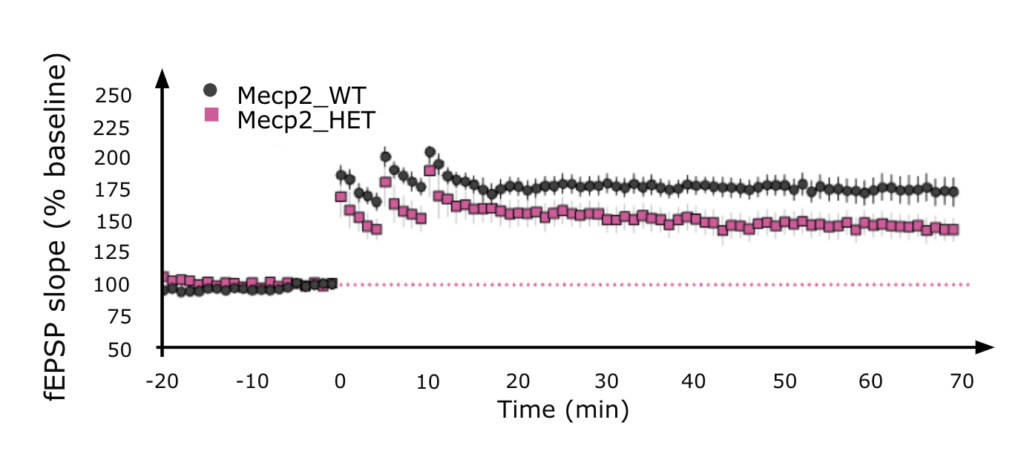
Differences in Long Term Potentiation (LTP)
Time course of responses recorded in CA1 area of hippocampus prior to and following LTP induction in brain slices from 6 month old female Mecp2 mice using HFS. A deficit in LTP is observed in the female Mecp2 HET mice compared to WT mice.
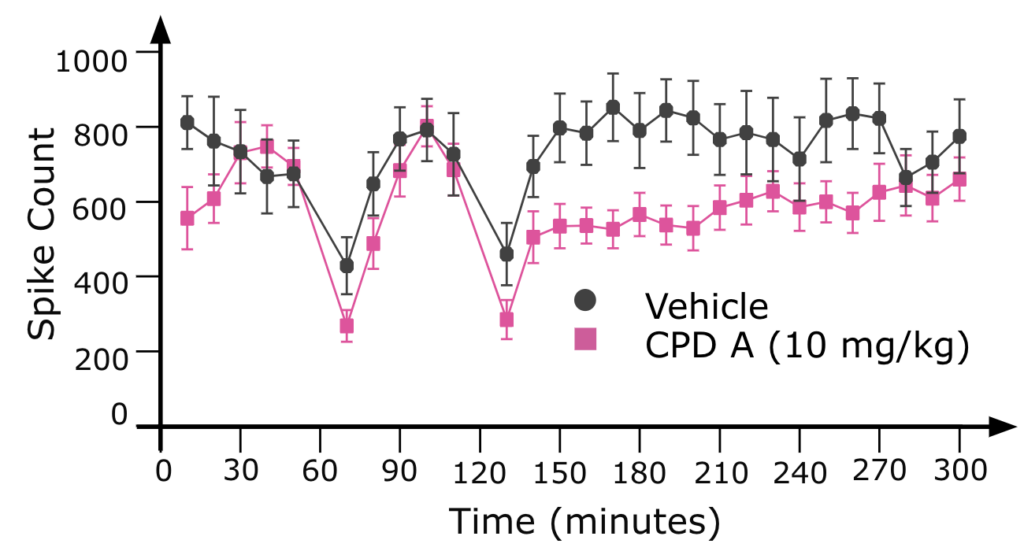
Seizure Activity and Rescues
EEG in 32-week old female Mecp2 HET mice show seizure activity that was ameliorated with Compound A.